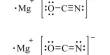Different definitions exist for “pharmaceutical analysis”, but it may be reasonably stated as the application of a process or series of processes in order to identify and/or quantify a drug or medicinal substance, the components of a solution or mixture, or the determination of the structures of chemical compounds.
“Pharmaceutical analysis” is the subject which deals mainly with the quantitative analysis of those chemicals and dosage forms associated with the practice of pharmacy. It provides training ground for the accuracy expected from pharmacy graduates, with the help of such compendia as the USP, BP, NF, IP etc.
In this aspect, it can be said that the scientist have given drugs to the people of the world and the pharmacy graduates guarantied the quality of those drugs now and in the future.
1.1.1 Objectives of analysis
The objective and purpose of the analysis has to be sensibly assessed before selecting an appropriate procedure. There are two objectives and/or purpose of analysis; including:
- First, it is one way to judge quality
- Second, any hazards associated with the drug can be ascertained and the necessary corrections to be made before the product is marketed
1.1.2 Types of analysis
There are two types of analysis;
- Qualitative analysis, and
- Quantitative analysis
The analytical procedure that deals with the qualitative property of a drug substance is called “qualitative analysis”. For example, when a completely unknown sample is presented to an analyst, the first requirement is usually to ascertain what substances are present in it. The fundamental problem may sometimes be encountered in modified form of deciding what impurities are present in a given sample, or perhaps of confirming that certain specified impurities are absent. The solution lies within the province of qualitative analysis.
The analytical procedure that deals with the quantitative measurements of a drug substance is called “quantitative analysis”. The main techniques employed in quantitative analysis based on:
- The quantitative performance of suitable chemical reactions and either measuring the amount reagent needed to complete the reaction, or ascertaining the amount of reaction product obtained
- Appropriate electrical measurements
- The measurements of certain spectroscopic properties
- The characteristic movement of a substance through a defined medium under controlled conditions
1.1.3 Key features of analysis
- Analyzing process should be carried out with an appropriate sample, which represents the whole material called a representative sample
- Analysis should be carried out with an appropriate methodology (1)
1.1.4 Stages of analysis
A complete chemical analysis, even for a single substance, involves a series of steps and procedures. Each one of them has to be carefully considered and assessed in order to minimize errors and to maintain accuracy and reproducibility. The steps are listed in the following:
Table 1.1.4.1: Stages of analysis
| Stages | Examples of procedure |
| Depends on the size and physical nature of the sample |
| Reduction of particle size, mixing for homogeneity, drying, determination of sample weight or volume |
| Heating, ignition, fusion, use of solvent(s), dilution |
| Filtration. extraction, ion exchange, chromatographic separation |
| Standardization, calibration, optimization, measurement of response; absorbance |
| Calculation of analytical result(s) for the sample, & statistical evaluation of data |
| Print out, data plotting, storage (archiving) |
1.1.5 Factors affecting the choice of analytical method
- The type of analysis required: elemental or molecular, routine or occasional.
- Problems arising from the nature of the material to be investigated, e.g. radioactive substances, corrosive substances, substances affected by water
- Possible interference from components of the material other than those of interest
- The concentration range to be investigated
- The accuracy required
- The facilities available, particularly the instruments
- The time required to complete the analysis
- The number of analysis of similar type which have to be performed
- Does the nature of the specimen, the kind of information sought, or the magnitude of the sample available indicate the use of non-destructive methods as opposed to the more commonly applied destructive methods? (2)
1.2 Classification of Official Assay Methods:
- Titrimetric Methods
A. Acid-Base Reactions1. Direct Titrations
a. Titration of an acid by a base
I. Titration of a liberated acid
II. Sorenson-Formal titration
III. Nonaqueous titration
b. Titration of a base by an acid
I. Titration of metal salts
II. Nonaqueous titration
III. Nonaqueous titration-Pifer-Wollish reagent
c. Kjeldhal Determination
2. Residual Titrations
a. Titration of excess acid by a base
I. After distillation of a volatile base
II. After addition to carbonate residues
III. After acylation reactions
IV. Nonaqueous titration
b. Titration of excess base by an acid
I. After saponification of an ester
II. After hydrolysis of an alkoxyl group
III. After distillation of a volatile base
B. Precipitation Reactions
1. Direct Titrations
a. With silver nitrate
b. With thiocyanate
I. Of theophyline-silver compound
II. Of halogen
III. Of mercury
IV. Of silver
c. Of a halogen with a mercuric ion
d. Of a halogen with a thorium (IV)
e. Of a liberated nitric acid
f. Of thiol with mercuric ion
2. Residual Titrations
a. With thiocyanate
I. Of theophyline-silver compound
II. Of silver
C. Redox Reactions
1. Direct Titrations
a. Involving ceric sulfate or ceric ammonium nitrate
b. Involving potassium permanganate
I. Using ferric alum and potassium permanganate
c. Involving dichlorophenol-indophenol
d. Involving potassium dichromate
e. Involving ferrous ammonium sulfate
f. Involving titanium trichloride
g. Involving ferric chloride
h. Involving standard bromide
i. Involving potassium ferricyanide
j. Involving sodium nitrate
k. With iodine
l. Involving iodine and thiosulfate
I. Iodimetric determination of phenol
II. Titration of iodine liberated from potassium iodide
III. Reaction of potassium iodide with excess periodate
m. of iodine with arsenite
n. involving potassium iodate
o. with thiosulfate
2. Residual Titrations
a. Of excess standard iodine
b. Of excess thiosulfate with iodine
c. Of generated iodine with thiosulfate
d. Of residual titanium with iron (III)
e. Of residual oxalic acid by potassium permanganate
f. Of residual iodine by sodium thiosulfate
D. Complaxometric Titration
1. Direct Titrations
a. With EDTA
b. With miscellaneous titrant
2. Residual Titrations
a. With EDTA
b. With metal ion
E. Large Anion Reagent and Large Cation Reagent Reactions
1. Titrations with sodium tetraphenylboron
2. Titration with sodium lauryl sulfate
3. Titration with tetra-n-butyl ammonium iodide
4. Titration with dioctyl sodium sulfosuccinate
Gravimetric Methods
A. Weighing Drug after Separation
B. Weighing a Derivative after Separation
C. Weighing a Residue after Ignition
Spectrometric Methods
A. Visible Absorption 1. Steroid
2. Dye-complex
3. Direct
4. Derivative formed
5. Starch-iodine reaction
B. Ultraviolet (UV) Spectrophotometric Absorption1. Direct
2. Derivative formed
3. Amphetamine
C. Infrared (IR) Absorption1. Direct
2. Derivative formed
D. Flame Photometric Emission
E. Flurometric Emission1. Native fluorescence
2. Fluorescent derivative formed
F. Atomic Absorption (AA)1. Flame
2. Furnace used
G. Nuclear Magnetic Resonance (NMR) Absorption1. Absolute method
2. Relative method
Electrochemical Methods
A. Voltammetry1. Polarography
2. Differential pulse polarography
3. Use of electrodes other than DME
B. Potentiometry
1. Ion-selective electrodes
Chromatographic Methods
A. Gas Liquid Chromatography (GLC)1. Direct assay
2. Derivative formed
B. High-Performance Liquid Chromatography (HPLC)
1. Direct assaya. Normal phase
b. Reverse phase
2. Derivative formeda. Normal phase
b. Reverse phase
C. Thin-Layer Chromatography (TLC)
1. Mobile phasea. Normal
b. Reverse
Miscellaneous Methods
A. Gasometric Assay
B. Assays involving Liquid Volume Measurements
C. Assays Involving Optical Rotation1. Direct
2. Derivative formed for assay
D. Assays Involving Specific Gravity
E. Assays of Radioactivity
F. Enzyme Assay
G. Proximate Assay1. Alkaloid assay
H. Biological Assay
I. Miscellaneous1. Fixed oils and waxes
J. Distillation
K. Functional Group Test
L. Vitamin Assays
M. Phase Solubility
N. Antibiotic Assays1. Microbial
2. Iodometric
3. Hydroxylamine
O. Individual components (3)
1.3 Ultraviolet (UV) Spectrophotometric Analysis
Spectrophotometric techniques are based on the absorption of light by an analyte or sample. The spectrophotometer measures the intensity amount of light transmitted through a transparent sample at a given wavelength and converts this number to an absorbance value. Absorbance is expressed as follows:
A=log(I0/I)
where I0 is equal to the intensity of light in the absence of the analyte and I is equal to the intensity of light in the presence of the analyte. In this calculation it is assumed that all of the light not transmitted to the detector is absorbed by the compounds to be measured in solution. However, two other possibilities exist. Either solids within the solution are scattering the light or the container for the solution is absorbing the light. For these reasons it is important to carefully filter soil samples and to always use a clean cuvette
In order for the substance to absorb certain wavelengths it is necessary for the given species to absorb light. The sample may appear colored and absorb in the visible or it may abosrb in the UV and thus appear colorless. By measuring each sample at the particular wavelength where the analyte absorbs light, a plot of the absorbance vs. the molarity of the solution can be created. Because the amount of radiant energy absorbed is proportional to the concentration of the absorbing material, it is possible to create a linear plot of absorbance vs. molarity when a set of standards of known concentration is measured. This is termed a Standard Curve. An equation for this line can be derived and the concentrations of phosphates in an unknown can then be quantitatively determined.
If the sample you are measuring absorbs in the visible range corresponding to one of the diodes on the Vernier Colorimeter, you can use these with the Logger Pro software as you did for the equilibrium/spectrophotometry experiment earlier this semester. If the sample absorbs in the UV, then you will need to use the Ocean Optics spectrophotometer and software.
To make a spectrophotometric analysis, you must have a series of standards which span the concentration range you expect in your samples that also have absorbance values less than 1. Once you make the measurements for the standards you will generate a standard curve to determine the concentration of the analyte in your samples.
1.3.1 Advantages of Spectrophotometric Analysis
- The precision (0.1%) is sometimes better than most instrumental methods
- Methods are usually superior to instrumental techniques for major component analysis
- It can perform within a few minutes
- It give us accurate result of different products
- When the sample throughout is large, e.g. for one-off analysis, spectrophotometric analysis are often preferable
- They are often used to calibrate and/or validate routine analysis using instruments
- The methods can be automated
1.3.2. Disadvantages of Spectrophotometric Analysis
- When the sample throughout is small, e.g. for one-off analysis, simple titrations are often preferable
- Unlike instrumental methods, the equipment must be require constant recalibration
- Methods are relatively expensive with high unit costs per determination
- This process is very much cost expensive
1.3.3 Method used in this project: Spectrophotometric Analysis
A Spectrophotometric analysis is a method in chemistry that allows quantitative analysis of the concentration of an unknown sample solution. It makes use of the measurement of the potency of different product and several raw materials. A Spectrophotometric analysis can be performed with any product and raw materials combination. (4)
1.4 Purpose of this study
Quality control of any pharmaceutical preparation is a very important aspect. No pharmaceutical products can be manufactured without any compounding or manufacturing error as no machine can provide hundred percent recovery results. However, there are various specifications that specify the considerable range of variations for the maintenance of product quality. The ultimate purpose of this study was to check out the product quality of Tetracycline HCl in terms of drug content by estimating the content of active Tetracycline hydrochloride from the capsule preparations which are available in the local market, to find out whether they meet the label claimed or not.
1.5 Literature Review
Antibiot Khimioter. 1991 Aug;36(8):18-20.
Spectrophotometric analysis of tetracycline hydrochloride in the presence of thiamine bromide and riboflavin
Spectrophotometric determination of tetracycline hydrochloride concentrations in the presence of thiamine bromide and riboflavin at a light absorption wave length of 300 nm was shown possible. When the ratio of tetracycline hydrochloride and the total of thiamine bromide and riboflavin (w/w) were at least 10:1 the error of the method was less than 2 per cent. The procedure is useful in quality control of the dosage forms.
CHEMICAL & PHARMACEUTICAL BULLETIN .Vol. 54 (2006) , No. 5 711
PARAFAC and PLS Applied to Spectrophotometric Determination of Tetracycline in Pharmaceutical Formulation and Biological Fluids
A simple and rapid analytical procedure was proposed for determination of tetracycline in pharmaceutical formulation, urine and plasma based on chemometrics methods and spectrophotometric measurements. The calibration set was constructed with twenty solutions in concentration range 0.25—13.00 μg ml−1 for tetracycline. The procedure was repeated at nine different pH values. Partial least squares (PLS) models were built at each pH and used to determinate a set of synthetic tetracycline solutions. The best model was obtained at pH 8.00 (PLS-PH8). Parallel factor analysis (PARAFAC) model was applied to a three-way array constructed using all the pH data sets and enabled better results. The capabilities of the method for the analysis of real samples were evaluated by determination of tetracycline in pharmaceutical formulations and biological fluids with satisfactory results.
| Key words | tetracycline; partial least squares (PLS); parallel factor analysis (PARAFAC); determination; pharmaceutical formulation; biological fluid |
J. Agric. Food Chem., 55 (13), 4973 -4979, 2007. Web Release Date: June 6, 2007
A sensitive and specific method is described for the simultaneous determination of oxytetracycline, tetracycline (TC), and chlortetracycline residues in edible swine tissues, by combining liquid chromatography with spectrofluorometric and mass spectrometry detection. The procedure involved a preliminary extraction with EDTA-McIlvaine buffer acidified at pH 4.0, followed by solid-phase extraction cleanup using a polymeric sorbent. The liquid chromatography analysis was performed with spectrofluorometric detection after postcolumn derivatization with magnesium ions. The limits of quantification were 50 g/kg for muscle and 100 g/kg for kidney tissues. The recovery values were greater than 77.8% for muscle and 65.1% for kidney. The method has been successfully used for the quantification of tetracyclines in swine tissues samples. The selective liquid chromatography mass spectrometric analysis for confirmation of oxytetracycline in one positive swine muscle sample was made by atmospheric pressure chemical ionization (APCI). The APCI mass spectra of the TCs gave the protonated molecular ion and two typical fragment ions, required for their confirmation in single ion monitoring scan mode in animal tissues.
Journal of Chromatography A ,Volume 1100, Issue 2, 30 December 2005, Pages 193-199
A simple, rapid, and simultaneous analysis method for oxytertracycline, tetracycline, chlortetracycline, penicillin G, ampicillin, and nafcillin in meat has been developed by using electrospray ionization tandem mass spectrometry. The sample preparation was performed by homogenizing with water followed by a centrifugal ultrafiltration, after addition of internal standards (demeclocycline, penicillin G-d5, ampicillin-d5 and nafcillin-d6). The MS/MS analysis involves the combined use of sample enrichment on the short column and a multiple reaction monitoring technique. The overall recoveries from animal (bovine and swine) muscle, kidney, and liver fortified at the levels of 0.05 and 0.1 ppm ranged from 70 to 115% with the coefficients of variation ranging from 0.7 to 14.8% (n = 5). Analysis time, including sample preparation and determination, is only 3 h per eight sample and detection limits for all antibiotics are 0.002 ppm. The method is considered to be satisfactory for the rapid screening of the tetracycline and penicillin antibiotic residues in meat.
A physicochemical study of the tetracycline coordination to oxovanadium(IV)
The interaction of tetracycline and oxovanadium(IV) in aqueous solution was studied by potentiometric and spectrophotometric methods. Oxovanadium(IV) ions form both a positively charged 1:1 and a neutral 2:1 metal–ligand complex with tetracycline. When a 1:1 ligand-to-metal ratio mixture is used at about pH 4.5 the 1:1 species predominates, being replaced at pH 6 by the binuclear complex. The binuclear complex has been isolated and fully characterised. Infrared and EPR studies suggest the existence of two distinct vanadyl binding sites. Our results indicate that the first vanadium coordinates to the BCD-ring system and the second one to the A-ring. Biological implications of the existence of a neutral complex at physiological pH are briefly discussed.
Author Keywords: Tetracycline; Oxovanadium(IV); Spectroscopy; Metal–ligand interaction
Stability studies of tetracycline in methanol solution.
The stability of tetracycline in methanol solution was investigated by UV–visible spectroscopy, HPLC and TLC methods. After dissolution in methanol, tetracycline decomposed rapidly under the influence of light and atmospheric oxygen, forming more than fourteen different degradation products. None of the previously reported degradation products, such as the epi- and anhydro-compounds, were detected as the final degradation products. The molecular structures for eight of the compounds were suggested by their product-ion mass spectra. A degradation sequence was proposed for the reactions of tetracycline with methanol. A new HPLC–MS mobile phase was developed, which solved the clogging problem at the interface between the HPLC and MS chamber and enabled a high separation efficiency.
Author Keywords: Tetracyclines; Antibiotics; Methanol