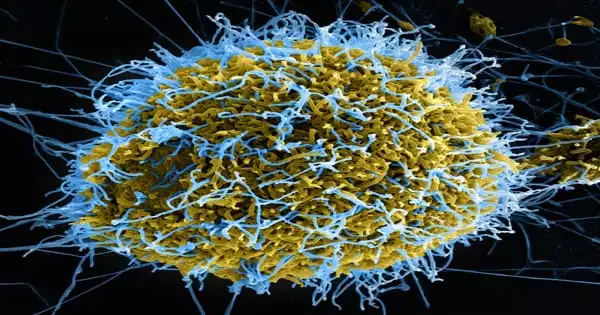
The first biologically accurate computer model of the HIV-1 viral liposome has been completed. The creation of sphingomyelin and cholesterol-rich microdomains is a key discovery from the simulations. HIV-1 is known to preferentially bud from areas of the host cell membrane with high concentrations of these components. Scientists anticipate that this basic research into viral envelopes will aid efforts to produce new HIV-1 therapies while also setting the groundwork for research into other enclosed viruses such as the novel coronavirus.
When is a container not just a container?
A double layer of fatty molecules known as lipids not only serves as a container for the HIV-1 virus, but it also plays an important function in the virus’s reproduction and infectivity. Scientists used supercomputers to create the first medically accurate computer model of the HIV-1 virus liposome, including its whole spherical lipid bilayer.
Furthermore, this research follows the publication of a novel atomistic model of the HIV-1 capsid, which carries the virus’s genetic material. The researchers anticipate that this basic research into viral envelopes will aid in the development of new HIV-1 therapies while also setting the groundwork for future research into other enclosed viruses such as the novel coronavirus, SARS-CoV-2.
“This work represents a full-scale investigation of the HIV-1 liposome with an unprecedented amount of molecular complexity,” said Alex Bryer, a Ph.D. student in the Perilla Laboratory at the University of Delaware’s Department of Chemistry and Biochemistry. Bryer is the lead author of the liposome-modeling study, which was published in the journal PLOS Computational Biology.
The scientists created a complicated chemical model of the HIV-1 liposome, which showed critical aspects of the liposome’s asymmetry. Most of these models assume a geometrically homogeneous structure and fail to account for the asymmetry inherent in biological containers.
Lipid Flip-Flop
Bryer and his colleagues explored a phenomenon known popularly as “lipid flip-flop,” which occurs when lipids in one of the bilayer’s leaflets are shifted or transported to the other leaflet. The leaflets flip-flop the lipids and interchange the molecules for a variety of reasons, including achieving dynamic equilibrium.
“For the spherical vesicle model of the liposome, our simulations show that asymmetry occurs spontaneously even without embedded proteins, and the vesicle can flip-flop to maintain an asymmetric composition within tight tolerances – even over biological timescales in excess of five microseconds,” Bryer said.
Interestingly, the science team did not observe the incidence of flip-flops in a flat membrane system, which suggests that the curvature of the envelope is intimately related to this biological process. “Nothing like this has ever been simulated before.” said study co-author Juan R. Perilla, an assistant professor in the Department of Chemistry and Biochemistry, University of Delaware.
“What was surprising for us is this dynamic equilibrium that the vesicle shows,” Perilla added. “Lipids are moving in and out, but the overall composition is not changing — that was surprising.”
It is believed that gp41 localizes to these domains. We demonstrated that these microdomains can develop in the vesicle without the assistance of proteins. They appear to appear out of nowhere.
Alex Bryer
Key Asymmetry
This important discovery demonstrates that the HIV-1 virus’s complex, asymmetric membrane composition can result in macroscopic features such as differential displacement of leaflets and the development of lipid microdomains. This development could have an impact on how membrane proteins interact with the membrane and carry out duties such as attaching to host cells and allowing the virus to enter them.
Microdomains are known to occur in HIV-1 and serve as a target for the localization of membrane proteins. One protein in particular, gp41, is required for membrane fusion, which is the process by which HIV-1 attaches to the host cell membrane and infects it.
“It is believed that gp41 localizes to these domains,” Bryer explained. “We demonstrated that these microdomains can develop in the vesicle without the assistance of proteins. They appear to appear out of nowhere.” This discovery could also explain why HIV-1 viruses preferentially budding, without the need for embedded proteins to mediate the creation of the microdomains that allow budding.
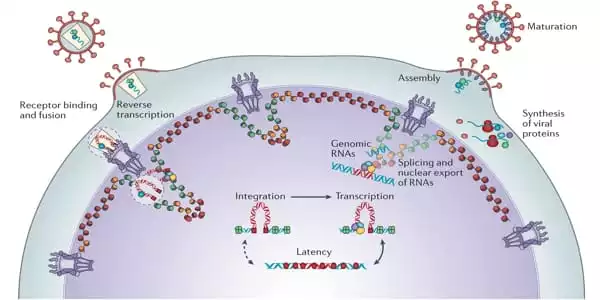
Supercomputer Simulations
Bryer and colleagues’ computer model has a diameter of 150 nm and is made up of 24 separate chemical ingredients. To represent a living environment, more than 300,000 lipid molecules are dissolved in water and ionized with sodium chloride. The scientists used the MARTINI coarse-grained model, which allowed them to minimize the degrees of freedom in the molecular system and perform simulation sampling on microsecond timeframes.
XSEDE, which is financed by the National Science Foundation, provided the scientists with supercomputer allocations and training. They employed the Stampede2 system at the Texas Advanced Computing Center (TACC) and the Bridges-2 system at the Pittsburgh Supercomputing Center through XSEDE (PSC). Additionally, they used Grizzly at the Los Alamos National Laboratory; Blue Waters at the National Center for Supercomputing Applications; and the Frontera system at TACC.
“Our study wouldn’t have been possible without XSEDE resources,” Bryer said. “We can achieve some very high sampling efficiencies using Stampede2 Skylake nodes, both to run the simulations and perform analyses.”
“I was able to perform calculations, and without needing to transfer data, I could set up a visualization session through the TACC portal and analyze and work with my data directly on Stampede2. That’s amazing,” added Bryer. He found that not having to transfer terabytes of data into a separate visualization computer node was “just huge in terms of productivity.” “We also used quite a bit of the high memory nodes on Bridges-2 of PSC,” Perilla said. They helped power simulations that compared the control, a flat HIV-1 viral membrane, to the curved one in dynamic equilibrium.
Furthermore, the Perilla Lab has moved the simulation work to their local cluster, the University of Delaware’s XSEDE-allocated DARWIN system. “It is critical to emphasize that XSEDE does more than just give incredibly valuable resources. There is training available, as well as additional options such as workshops “According to Perilla.
“I had never logged into a supercomputer before joining the club,” Bryer explained. He remembers receiving important instruction in XSEDE workshops on OpenMP, MPI, and OpenACC, which help scientists parallelize their computer code.
Frontera Work
Bryer also emphasized the analysis work performed on TACC’s Frontera, the world’s fastest academic supercomputer. “A lot of the analyses in the manuscript were made possible by parallel I/O using Luster,” Bryer explained. “We were able to swiftly categorize the volume surrounding the vesicle using Frontera and process our data in minutes. We predicted that running the study in a serial naive implementation would take roughly three weeks.”
All of the computing power and experience at the Perilla Lab has been directed toward unraveling the riddles of what occurs to the HIV-1 viral envelope during infection.
“While this study does not provide the whole answer, it’s getting there in what the lipids are doing and what integral membrane proteins are doing or could be doing; and not only how proteins like gp41 interact with human receptors but also how they transmit their signals and how that is related to lipid composition,” Perilla said.
“This computational study provides an opportunity for drug development research,” Perilla added. Since lipid symmetry is maintained by the curvature of the envelope, a promising possibility yet unexplored is development of small molecules that affect the symmetry and potentially yield a therapeutic target.
HIV-1 Capsid
Prior to the liposome study, Perilla and colleagues made history by creating the first-ever atomistic model of the HIV-1 capsid, the envelope for its genetic material, in the presence of the metabolite IP6 using supercomputers. The findings were published in the journal Science Advances. It also made use of the XSEDE-allocated Bridges-2 and Stampede2 supercomputers.
The calculations, which were corroborated by cryo-electron tomography data, revealed that IP6 might attach to the capsid in two sites, rather than only one, as previously supposed. This discovery is significant because the capsid is exposed to the cytoplasm during infection and must pass through the nuclear import mechanism, especially the nuclear pore complex. All these pieces together point to the capsid being able to “sense” in an as yet unknown way the concentration of IP6.