Influenza, sometimes known as the flu, is a viral respiratory infection caused by influenza viruses. These viruses can undergo mutations, which play an important role in the evolution of the influenza virus. Mutation accessibility refers to the ease with which certain alterations can occur in the viral genome.
The influenza (flu) virus is constantly evolving and adapting by gaining new mutations. Scientists at St. Jude Children’s Research Hospital have added a new layer of insight to explain why and how flu viruses develop. The “survival of the accessible” concept supplements the more widely accepted “survival of the fittest” paradigm of evolution. The study was published today in Science Advances.
Viruses evolve quickly as a result of continual genetic changes. Because of this rapid fluctuation, people get flu shots every year to combat the latest flu variation that has emerged as the prevalent strain. These mutations are frequently observed in the framework of traditional evolutionary thought, in which variation fitness determines which modified virus emerges as the dominant strain in a population. The St. Jude team studied this notion and defined an alternative evolutionary principle, which they say is a significant driver of evolution, dubbed “variant accessibility.”
The research, led by Alexander Gunnarsson, Ph.D., and M. Madan Babu, Ph.D., St. Jude Department of Structural Biology and Center of Excellence for Data-Driven Discovery, involved creating a model of mutational accessibility to help predict how and why specific mutations emerge in a population during viral evolution.
Our model can be used to predict whether a specific type of mutation is likely to emerge as a tumor driver or as a treatment-resistant mutation. We hope that our work will stimulate research into the mutational biases that drive viral and tumor evolution.
M. Madan Babu
The unappreciated role of variant accessibility
The genomic alphabet only has four letters representing the nucleotides: (A)denosine, (T)hymine, (G)uanine, and (C)ytosine. Groups of three nucleotides within a protein-coding gene are called a codon. Codons act like a recipe for assembling proteins, encoding for a specific amino acid. Mutations occur when nucleotides are altered, for instance, during replication. This alteration leads to a different amino acid being used to make the protein. But not all mutations are equally likely to emerge, as Babu and Gunnarsson discovered.
“The process of genetic replication has inherent biases built in, such as the relative ease of an A to be mutated to a C rather than to a G,” Babu explained. “This means that the pool of mutants with this A-to-C mutation is larger, and surviving variants will predominantly emerge from that particular pool, even though there may be a fitter sequence with an A-to-G mutation.”
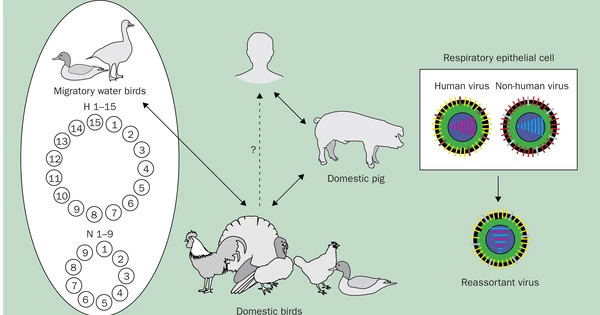
Gunnarsson and Babu used the influenza virus as a case study to transfer this concept into a mathematical model. Based on the accessibility of a mutation, their model allows researchers to anticipate the route of future evolution. Exploring how individual protein locations can gain or lose the ability to be changed after a mutation was of special interest. They then investigated how the protein’s function was affected by this gain or loss.
Phosphorylation is one type of modification. It happens when a phosphate molecule is attached to certain amino acids in a protein. In the case of the flu, phosphorylation can assist the virus in hijacking the host molecular pathways and successfully infecting the host. Such mutations may have been critical to influenza pandemics of the past, and it is these datasets that Gunnarsson and Babu used to develop their model.
The importance of jackpot events
The model also helped the researchers better understand a long-conceptualized mutation property, the jackpot event. These are mutations that occur by chance early in the growth of a population, leading to a continuous benefit seen throughout the descendants. “The more accessible a genotype is, the more frequent these specific jackpot events are because it’s simply a probabilistic event,” Gunnarsson explained. “If a particular gene is a hundred times more likely to acquire a specific mutation, you’ll see that jackpot event happening proportionately more frequently. These events are important in evolution and are driven primarily by how accessible the variants are.”
More accessible mutations are likely to be predominant in a population even though they may not be the fittest mutation. “If the probability of acquiring the fittest mutation is one out of hundreds of trillions,” Gunnarsson said, “the likelihood of it reaching fixation in a population, even if it’s the fittest mutation, is low. When you have multiple instances of jackpot mutations happening, statistically, the prevalence of this variant increases massively, even if it’s less fit compared to another, more fit but less accessible mutant.”
Furthering our understanding of mutational bias and predicting outcomes in evolving systems
The concept of variant accessibility is simple in its simplicity, but it is a balancing of statistical probability, as are most things in nature. From the mutation event and differences in the frequency of various nucleotide modifications to codon redundancy (several codons for the same amino acid), evolutionary routes are driven by a complex balance of components.
“Furthering our understanding of biochemical mutational biases (e.g., during replication) in viruses can open up new directions and possibilities because it’ll give much better insights into how a virus is likely to evolve,” Babu explained. In fact, the model is being used to historical data on how the flu virus has changed within the context of mutational accessibility in order to better correctly forecast viral evolution.
The ability to predict viral evolutionary outcomes based on accessibility has piqued the interest of influenza expert Richard Webby, Ph.D., of St. Jude Department of Host-Microbe Interactions and Director of the World Health Organization Collaborating Centre for Studies on the Ecology of Influenza in Animals and Birds.
“There are many scenarios in public health where we try and predict the evolutionary path of influenza viruses, including selecting the most appropriate vaccines for future influenza,” Webby explained. “The ‘survival of the accessible’ model will strengthen these predictions and allow us to more confidently identify viruses that are more likely to take on worrying traits.”
This approach extends beyond influenza and virology, directing future study into mutational biases in various diseases. In cancer, for example, the model can assist solve a variety of pathological problems, such as why certain cancer-causing or drug-resistance mutations appear repeatedly.
“Our model can be used to predict whether a specific type of mutation is likely to emerge as a tumor driver or as a treatment resistance mutation,” Babu said. “We hope that our work will stimulate research into the mutational biases that drive viral and tumor evolution.” We will be able to forecast mutational outcomes in evolving genetic systems if we can measure and better understand the biochemical processes that contribute to mutational bias. The ability to forecast consequences before they occur will help us to be prepared when they do occur.”