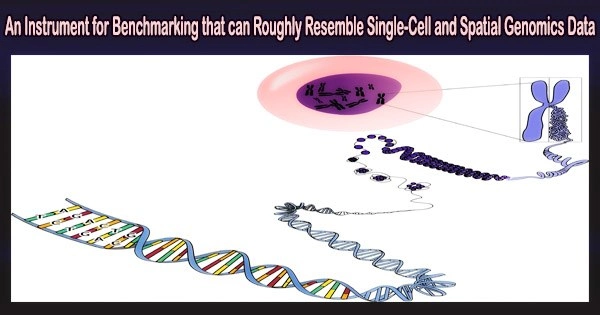
A benchmarking tool for medical and biological researchers who use cutting-edge technologies to study diseases and potential treatments has been created by UCLA researchers using a “all-in-one,” next-generation statistical simulator that can take in a variety of data to produce realistic synthetic data.
In particular, the new “in silico” or computer-modeling system can assist researchers in assessing and validating computational approaches.
The basis for examining the genetic make-up (genome-wide gene expression levels) of cells is single-cell RNA sequencing, also known as single-cell transcriptomics.
In recent years, spatial transcriptomic technologies have made it possible to profile gene expression levels with spatial location information of cell “neighborhoods,” showing precise locations and movements of cells within tissue. Additional “omics” have provided detail on a variety of molecular features.
“Thousands of computational methods have been developed to analyze single-cell and spatial omics data for a variety of tasks, making method benchmarking a pressing challenge for method developers and uses,” said Jingyi Jessica Li, Ph.D., a UCLA researcher and professor in statistics, biostatistics, computational medicine, and human genetics.
The system’s transparent modeling and interpretable parameters can help users explore, alter, and simulate data. Overall, scDesign3 is a multi-functional suite for benchmarking computational methods and interpreting single-cell and spatial omics data.
Jingyi Jessica Li
Li is also affiliated with the Gene Regulation research area at the UCLA Jonsson Comprehensive Cancer Center. Li leads a research group titled the Junction of Statistics and Biology.
“Although simulators have evolved and become more powerful, there are numerous limitations. Few can generate realistic single-cell RNA sequencing data from continuous cell trajectories by mimicking real data, and most lack the ability to simulate data of multi-omics and spatial transcriptomics. We introduced the scDesign3, which we believe is the most realistic and versatile simulator to date, to fill the gap between researchers’ benchmarking needs and the limitations of existing tools,” said Li, senior author of a study published May 11 in Nature Biotechnology.
The UCLA researchers say they believe scDesign3 “offers the first probabilistic model that unifies the generation and inference for single-cell and spatial omics data. Equipped with interpretable parameters and a model likelihood, scDesign3 is beyond a versatile simulator and has unique advantages for generating customized in silico data, which can serve as negative and positive controls for computational analysis, and for assessing the goodness-of-fit of inferred cell clusters, trajectories, and spatial locations in an unsupervised way.”
Goodness-of-fit is a measure of how well a statistical model fits a set of observations.
According to the authors, the system’s “transparent modeling and interpretable parameters can help users explore, alter, and simulate data. Overall, scDesign3 is a multi-functional suite for benchmarking computational methods and interpreting single-cell and spatial omics data.”