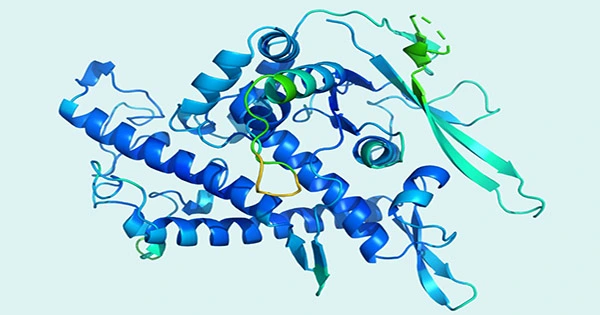
Without the protein molecules that support vital biological functions including photosynthesis, enzymatic degradation, sight, and our immune system, life on Earth would not exist as we know it. And like other aspects of nature, humankind is still learning about all the different kinds of proteins that are actually out there. The ESM Metagenomic Atlas, a first-of-its-kind metagenomic database, was created by Meta researchers instead of scouring the planet’s most inhospitable regions in search of novel microorganisms that might possess a new type of organic molecule. This database has the potential to be 60 times faster than current protein-folding AI performance.
The name “metagenomics” is really a coincidence. The study of “the structure and function of complete nucleotide sequences extracted and studied from all the organisms (usually microorganisms) in a bulk sample” is a relatively new but very real field of science. These techniques, which work similarly to gas chromatography in that you’re seeking to determine what’s there in a certain sample system, are frequently used to detect the bacterial communities residing on our skin or in the soil. The issue is that even though advances in genomics have identified the sequences for a large number of novel proteins, simply knowing those sequences does not explain how they fit together to form a functional molecule, and it can take anywhere from a few months to a few years to figure it out experimentally. as each molecule. No one has time for that.
The NCBI, the European Bioinformatics Institute, and the Joint Genome Institute all launched similar databases that have already compiled billions of previously unknown protein structures. According to a press statement issued by Meta on Tuesday, the company is providing “a revolutionary protein-folding approach that harnesses huge language models to generate the first comprehensive understanding of the structures of proteins in a metagenomics database at the scale of hundreds of millions of proteins.”
The Meta research team stated on Tuesday that the ESM Metagenomic Atlas “would enable scientists to examine and evaluate the structures of metagenomic proteins at the scale of hundreds of millions of proteins.” Researchers can utilize this to find previously uncharacterized structures, look for ancient evolutionary connections, and find novel proteins that may have applications in medicine and other fields.
Like languages, proteins are composed of their constituent atoms, which you can combine in any way you like, but only when put together in a certain order will result in a functional molecule, or coherent thinking (a molecular sentence). Although the analogy isn’t exact, Meta’s system significantly enhances our ability to understand the syntax and grammar of organic chemistry. According to the rules of physics, molecules fold into complicated three-dimensional shapes, which are described by a protein’s sequence, the scientists said. Protein sequences include statistical patterns that reveal details about the folded structure of the protein, according to research.
In particular, Meta’s Evolutionary Scale Modeling AI uses masked language modeling, a type of self-supervised learning, to treat gene sequences like a game of Mad Libs for O-Chem. The research team stated, “We trained a language model using the sequences of millions of natural proteins.” “This method requires the model to accurately complete the blanks in a text passage, such as “To _ or not to __, that is the .” Using millions of different proteins, we trained a language model to fill in the blanks in a protein sequence like “GL KKE AHY G.”
ESM-2, the resulting “protein language model,” has 15 billion parameters and is the largest model of its kind to date. On a cluster of about 2,000 GPUs, the “new structure prediction capacity enabled us to predict sequences for the more than 600 million metagenomic proteins in the atlas in just two weeks.” Well, forget about months and years.