Huntington’s disease is a genetic disorder that affects the nerve cells in the brain, particularly the ones in the basal ganglia, which are responsible for controlling movement, balance, and coordination. The disease is caused by a mutation in the huntingtin gene, which produces an abnormal version of the huntingtin protein. This abnormal protein damages and kills nerve cells, particularly in the areas of the brain that control movement. As the disease progresses, it also affects other areas of the brain, including those involved in cognitive and behavioral function. This leads to a decline in cognitive and motor skills, as well as psychiatric symptoms such as depression and irritability.
Huntington’s disease affects two distinct cell populations in the striatum, according to neuroscientists. They believe that neurodegeneration in one of these populations causes motor impairments, whereas damage to the other population, which is located in a structure called the striosome, may account for the mood disorders seen in the early stages of the disease.
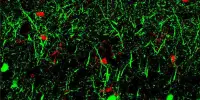
Neurons in the striatum, a part of the brain affected by Huntington’s disease, are among the most severely affected. The degeneration of these neurons contributes to patients’ loss of motor control, which is one of the disease’s major symptoms.
Neuroscientists at MIT have now shown that two distinct cell populations in the striatum are affected differently by Huntington’s disease. They believe that neurodegeneration of one of these populations leads to motor impairments, while damage to the other population, located in structures called striosomes, may account for the mood disorders that are often seen in the early stages of the disease.
As many as 10 years ahead of the motor diagnosis, Huntington’s patients can experience mood disorders, and one possibility is that the striosomes might be involved in these.
Ann Graybiel
“As many as 10 years ahead of the motor diagnosis, Huntington’s patients can experience mood disorders, and one possibility is that the striosomes might be involved in these,” says Ann Graybiel, an MIT Institute Professor, a member of MIT’s McGovern Institute for Brain Research, and one of the senior authors of the study.
Using single-cell RNA sequencing to examine the genes expressed in Huntington’s disease mouse models and postmortem brain samples from Huntington’s patients, the researchers discovered that as the disease progresses, cells of the striosomes and another structure, the matrix, lose their distinguishing features. The researchers hope that their mapping of the striatum and how it is affected by Huntington’s will lead to new treatments that target specific brain cells.
According to the researchers, this type of analysis could shed light on other brain disorders that affect the striatum, such as Parkinson’s disease and an autism spectrum disorder.
Myriam Heiman, an associate professor in MIT’s Department of Brain and Cognitive Sciences and a member of the Picower Institute for Learning and Memory, and Manolis Kellis, a professor of computer science in MIT’s Computer Science and Artificial Intelligence Laboratory (CSAIL) and a member of the Broad Institute of MIT and Harvard, are also senior authors of the study. Ayano Matsushima, a McGovern Institute research scientist, and Sergio Sebastian Pineda, an MIT graduate student, are the lead authors of the paper, which appears in Nature Communications.
Neuron vulnerability
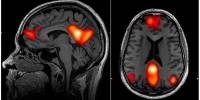
Huntington’s disease causes degeneration of brain structures known as basal ganglia, which are responsible for movement control and also play roles in other behaviors and emotions. Graybiel has been researching the striatum, a part of the basal ganglia involved in making decisions that require evaluating the outcomes of a specific action, for many years.
Graybiel discovered many years ago that the striatum is divided into striosomes, which are clusters of neurons, and the matrix, which surrounds the striosomes. She has also demonstrated that striosomes are required for making decisions that necessitate an agonizing cost-benefit analysis.
Richard Faull of the University of Auckland discovered in a 2007 study that striosomes showed significant degeneration in postmortem brain tissue from Huntington’s patients. Faull also discovered that many of the patients had symptoms of mood disorders such as depression prior to developing motor symptoms.
Graybiel collaborated with Kellis and Heiman to study the gene expression patterns of striosomal and matrix cells to further investigate the links between the striatum and the mood and motor effects of Huntington’s. To accomplish this, the researchers used single-cell RNA sequencing to examine human brain samples as well as brain tissue from two mouse models of Huntington’s disease.
Neurons in the striatum are classified as either D1 or D2 neurons. D1 neurons are part of the “go” pathway, which starts an action, whereas D2 neurons are part of the “no-go” pathway, which stops an action. D1 and D2 neurons can be found in both striosomes and the matrix.
The analysis of RNA expression in each of these cell types revealed that striosomal neurons are more vulnerable to Huntington’s disease than matrix neurons. Furthermore, D2 neurons are more vulnerable than D1 neurons within the striosomes.
Striosomal disorders
Damage to the striosomes, which are known to be involved in mood regulation, may be responsible for the mood disorders that affect Huntington’s patients in the early stages of the disease, according to the findings. Later on, degeneration of the matrix neurons likely contributes to the decline of motor function, the researchers say.
In the future, the researchers hope to investigate how striosome degeneration or abnormal gene expression may contribute to other brain disorders. Previous research has shown that striosome overactivity can result in the development of repetitive behaviors such as autism, obsessive compulsive disorder, and Tourette’s syndrome. According to the findings of this study, at least one of the genes discovered to be overexpressed in the striosomes of Huntington’s brains is also linked to autism.