Although the devastating and incurable Mad Cow disease and its human counterpart Creutzfeldt-Jakob disease have been absent from the news for years, scientists have not taken a break from their own work since prions-caused infectious diseases in mammals still affect mammals today.
Researchers from Brown University reveal an important new understanding of how prion proteins, the infectious agents, become transmissible in the Oct. 29 (2010) issue of Science: At least in yeast, the effectiveness of prion complexes in spreading is determined by their size rather than their quantity.
“The dogma in the field was that the misfolding of the protein is sufficient to cause disease, and the clinical course of the infection depended on the amplification of the misfolded protein,” said Tricia Serio, associate professor of molecular biology, cell biology and biochemistry.
“But over the years in mammals it has become clear that the abundance of misfolded protein is not a good predictor of disease progression. The question is, What else has to happen for you to get the clinical pathology?”
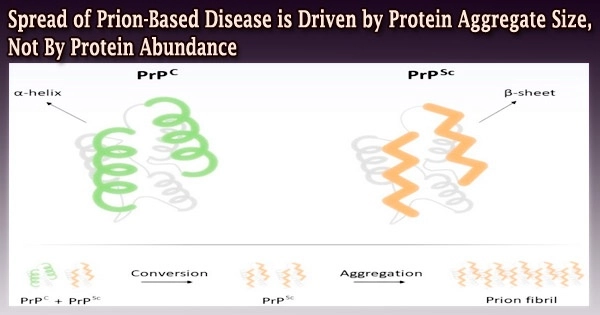
Although prion proteins are normally produced by cells, researchers are unsure of their typical function in mammals. These proteins collect into things called aggregates when they misfold in cells, while other proteins, called chaperones, work to disassemble the aggregates.
The shape or conformation that the prion protein has taken on determines the rates at which this assembly and disassembly happens.
The dogma in the field was that the misfolding of the protein is sufficient to cause disease, and the clinical course of the infection depended on the amplification of the misfolded protein. But over the years in mammals it has become clear that the abundance of misfolded protein is not a good predictor of disease progression. The question is, What else has to happen for you to get the clinical pathology?
Tricia Serio
“Different conformations of the same prion protein can dramatically alter the spread of pathology and the incubation time of prion diseases,” Serio said. “We wanted to learn how.”
The Brown team discovered that what influences a prions’ capacity to spread from cell to cell is the size of the structures into which they organize, according to Serio. This was done by integrating research in yeast cells with mathematical models.
The aggregates lose their ability to spread between cells when they grow too big. Prion aggregates that don’t grow in size transmit more effectively.
“In this paper we changed the transmissibility just by shifting the size,” Serio said. “We could change it in either direction.”
The evidence was easy to see. Under a microscope, postdoctoral researcher Aaron Derdowski saw that smaller prion aggregates fared better than bigger ones as they traveled between cells. He also monitored the genetic spread of various prion formations by examining afflicted cell populations.
Suzanne Sindi, a postdoctoral researcher with appointments in molecular biology, cell biology, and biochemistry as well as the Center for Computational Molecular Biology, modeled how cells create and spread prion aggregates in conjunction with the experimental work. She did this by running a novel simulation of the process on a computing cluster in the Center for Computation and Visualization at Brown.
The model where aggregate size rather than abundance was the determining factor best captured experimental findings.
Implications for disease
Serio says the insights the team has gained in yeast may better explain what others have observed in mammals as well.
“Previously it was not clear why you would have those outcomes,” she said.
In the end, the results might guide future plans for creating a prion infection treatment. Serio warned that if researchers created a therapy to prevent the creation of prion aggregates without understanding the significance of aggregate size, they would unintentionally make matters worse by generating smaller aggregates.
“A more effective strategy might be to control the size of the aggregates,” she said, “rather than their presence or absence.”
According to Serio, the findings may also be relevant to other neurodegenerative conditions involving misfolded proteins, such as Parkinson’s and Alzheimer’s illnesses.
Other authors on the paper include graduate students Courtney Klaips and Susanne DiSalvo. The National Institutes of Health funded the work.