The complex set of mechanisms that control gene expression in living organisms is referred to as gene regulation. It is crucial in determining the development, function, and adaptability of organisms. While our understanding of gene regulation has greatly advanced, there is still much to learn. Let us investigate a novel perspective on gene regulation based on current knowledge.
Researchers have demonstrated that they can map interactions between gene promoters and enhancers with 100 times higher resolution than was previously possible. Much of the human genome is made up of regulatory regions that control which genes are expressed within a cell at any given time. Those regulatory elements can be located near a target gene or up to 2 million base pairs away from the target.
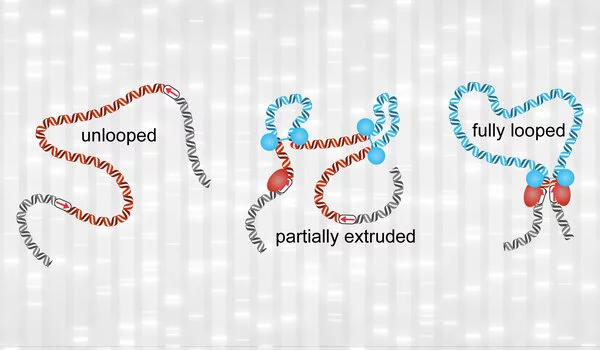
To facilitate these interactions, the genome loops itself in a 3D structure, bringing distant regions closer together. MIT researchers have demonstrated that they can map these interactions with 100 times higher resolution than was previously possible using a new technique.
Researchers can now affordably study the interactions between genes and their regulators, opening up a world of possibilities not just for us, but also for dozens of labs that have already expressed interest in our method
Viraat Goel
“Using this method, we generate the highest-resolution maps of the 3D genome ever generated, and what we see are a lot of previously unseen interactions between enhancers and promoters,” says Anders Sejr Hansen, the Underwood-Prescott Career Development Assistant Professor of Biological Engineering at MIT and the study’s senior author. “We are very excited to be able to reveal a new layer of 3D structure with our high resolution.”
The findings of the researchers suggest that many genes interact with dozens of different regulatory elements, though more research is needed to determine which of those interactions are most important to the regulation of a given gene.
“Researchers can now affordably study the interactions between genes and their regulators, opening up a world of possibilities not just for us, but also for dozens of labs that have already expressed interest in our method,” says Viraat Goel, an MIT graduate student and one of the paper’s lead authors. “We’re excited to bring the research community a tool that will help them disentangle the mechanisms driving gene regulation.”
MIT postdoc Miles Huseyin is also a lead author of the paper, which appears today in Nature Genetics.
High-zesolution mapping
Scientists believe that more than half of the genome is made up of regulatory elements that control genes, which account for only about 2% of the genome. Many variants that appear in these regulatory regions have been identified by genome-wide association studies, which link genetic variants to specific diseases. Determining which genes these regulatory elements interact with may aid researchers in understanding how diseases develop and, potentially, how to treat them.
To discover those interactions, researchers must first determine which parts of the genome interact with one another when chromosomes are packed into the nucleus. Chromosomes are organized into structural units called nucleosomes, which are strands of DNA tightly wound around proteins that help the chromosomes fit within the nucleus’s small confines.
Over a decade ago, a team led by MIT researchers developed Hi-C, which revealed that the genome is organized as a “fractal globule,” allowing the cell to tightly pack its DNA while avoiding knots. This architecture also allows the DNA to unfold and refold easily as needed. Hi-C involves using restriction enzymes to cut the genome into many small pieces and biochemically linking pieces that are close together in 3D space within the cell’s nucleus. They then use amplifying and sequencing to determine the identities of the interacting pieces.
While Hi-C reveals a lot about the overall 3D organization of the genome, its resolution is too low to detect specific interactions between genes and regulatory elements like enhancers. Enhancers are short DNA sequences that can help activate gene transcription by binding to the promoter of the gene, which is where transcription begins.
To achieve the resolution necessary to find these interactions, the MIT team built on a more recent technology called Micro-C, which was invented by researchers at the University of Massachusetts Medical School, led by Stanley Hsieh and Oliver Rando. Micro-C was first applied in budding yeast in 2015 and subsequently applied to mammalian cells in three papers in 2019 and 2020 by researchers including Hansen, Hsieh, Rando and others at University of California at Berkeley and at UMass Medical School.
Micro-C achieves higher resolution than Hi-C by using an enzyme known as micrococcal nuclease to chop up the genome. Hi-C’s restriction enzymes cut the genome only at specific DNA sequences that are randomly distributed, resulting in DNA fragments of varying and larger sizes. By contrast, micrococcal nuclease uniformly cuts the genome into nucleosome-sized fragments, each of which contains 150 to 200 DNA base pairs. This uniformity of small fragments grants Micro-C its superior resolution over Hi-C.
However, since Micro-C surveys the entire genome, this approach still doesn’t achieve high enough resolution to identify the types of interactions the researchers wanted to see. For example, if you want to look at how 100 different genome sites interact with each other, you need to sequence at least 100 multiplied by 100 times, or 10,000. The human genome is very large and contains around 22 million sites at nucleosome resolution. Therefore, Micro-C mapping of the entire human genome would require at least 22 million multiplied by 22 million sequencing reads, costing more than $1 billion.
To reduce the cost, the researchers devised a method for performing more targeted sequencing of the genome’s interactions, allowing them to focus on segments of the genome that contain genes of interest. By focusing on regions spanning a few million base pairs, the number of potential genomic sites is reduced by a thousandfold, and sequencing costs are reduced by a millionfold, to around $1,000. The new method, known as Region Capture Micro-C (RCMC), can thus generate maps 100 times richer in information than other published techniques at a fraction of the cost.
Many interactions
The researchers chose five regions ranging in size from hundreds of thousands to approximately 2 million base pairs for this study because of interesting features revealed by previous studies. Among these is Sox2, a well-studied gene that plays an important role in tissue formation during embryonic development.
After capturing and sequencing the DNA segments of interest, the researchers discovered numerous enhancers that interact with Sox2, as well as previously unknown interactions between nearby genes and enhancers. In other regions, particularly those densely packed with genes and enhancers, some genes interacted with as many as 50 other DNA segments, with each interacting site contacting an average of 25 others.
“People have seen multiple interactions from one bit of DNA before, but it’s usually on the order of two or three, so seeing this many of them was quite significant in terms of difference,” Huseyin explains.
The researchers’ technique, however, does not reveal whether all of those interactions occur at the same time or at different times, or which of those interactions is the most important. The researchers also discovered that DNA appears to coil itself into nested “microcompartments” that facilitate these interactions, but they couldn’t figure out how microcompartments form. Further research into the underlying mechanisms, the researchers hope, will shed light on the fundamental question of how genes are regulated.