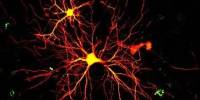
Researchers conducted the largest-scale examination of the genomic, epigenomic, and transcriptome changes that occur in every cell type in Alzheimer’s patients’ brains, using more than 2 million cells from over 400 postmortem brain samples.
Alzheimer’s disease affects about 6 million individuals in the United States, and there are very few FDA-approved medicines to reduce the illness’s progression. MIT researchers conducted the most comprehensive investigation yet of the genomic, epigenomic, and transcriptome changes that occur in every cell type in Alzheimer’s patients’ brains with the aim of uncovering novel targets for potential Alzheimer’s treatments.
The researchers examined how gene expression changes as Alzheimer’s disease proceeds using over 2 million cells from over 400 postmortem brain tissues. They also monitored changes in epigenomic alterations in cells, which aid in determining which genes are turned on or off in a given cell. These techniques, taken together, provide the most complete picture yet of the genetic and molecular basis of Alzheimer’s disease.
What we set out to do was blend together our computational and our biological expertise and take an unbiased look at Alzheimer’s at an unprecedented scale across hundreds of individuals – something that has just never been undertaken before.
Manolis Kellis
The findings are reported in a series of four studies published today in Cell. Li-Huei Tsai, director of MIT’s Picower Institute for Learning and Memory, and Manolis Kellis, a computer science professor in MIT’s Computer Science and Artificial Intelligence Laboratory (CSAIL) and a member of the Broad Institute of MIT and Harvard, conducted the research.
“What we set out to do was blend together our computational and our biological expertise and take an unbiased look at Alzheimer’s at an unprecedented scale across hundreds of individuals — something that has just never been undertaken before,” Kellis said.
The findings imply that genetic and epigenetic alterations interact to drive the clinical features of the disease. “It’s a multifactorial process,” explains Tsai. “These papers together use different approaches that point to a converging picture of Alzheimer’s disease where the affected neurons have defects in their 3D genome, and that is causal to a lot of the disease phenotypes we see.”
A complex interplay
Many drug development efforts for Alzheimer’s disease have focused on the amyloid plaques that form in patients’ brains. The MIT researchers aimed to identify alternative possible techniques in their latest series of studies by evaluating the disease’s molecular drivers, the cell types that are most vulnerable, and the underlying biological mechanisms that drive neurodegeneration.
The researchers conducted transcriptome and epigenomic analyses on 427 brain samples from the Religious Orders Study/Memory and Aging Project (ROSMAP), a long-term study that has studied memory, motor, and other age-related changes in older persons since 1994. There were 146 persons with no cognitive impairment, 102 with mild cognitive impairment, and 144 with Alzheimer’s-related dementia in these samples.
The researchers employed single-cell RNA-sequencing to assess the gene expression patterns of 54 types of brain cells from these samples in the first Cell study, which focused on gene expression changes, and found cellular processes that were most impaired in Alzheimer’s patients. Among the most notable were defects in the expression of genes essential in mitochondrial function, synaptic signaling, and protein complexes required to maintain genome structural integrity.
This gene expression study, which was led by former MIT postdoc Hansruedi Mathys, graduate student Zhuyu (Verna) Peng, and former graduate student Carles Boix, also found that genetic pathways related to lipid metabolism were highly disrupted. In work published in Nature last year, the Tsai and Kellis labs showed that the strongest genetic risk for Alzheimer’s, called APOE4, interferes with normal lipid metabolism, which can then lead to defects in many other cell processes.
The researchers studied gene expression patterns in patients with cognitive deficits and those who did not, including some who stayed bright while having some degree of amyloid deposition in the brain, a trait known as cognitive resilience, in the study conducted by Mathys. According to the findings, cognitively robust adults exhibited higher populations of two subsets of inhibitory neurons in the prefrontal cortex. These cells appear to be more sensitive to neurodegeneration and cell death in persons with Alzheimer’s disease-linked dementia.
“This revelation suggests that specific inhibitory neuron populations might hold the key to maintaining cognitive function even in the presence of Alzheimer’s pathology,” Mathys says. “Our study pinpoints these specific inhibitory neuron subtypes as a crucial target for future research and has the potential to facilitate the development of therapeutic interventions aimed at preserving cognitive abilities in aging populations.”
Epigenomics
In the second Cell paper, led by former MIT postdoc Xushen Xiong, graduate student Benjamin James, and former graduate student Carles Boix PhD ’22, the researchers examined some of the epigenomic changes that occurred in 92 people, including 48 healthy individuals and 44 with early or late-stage Alzheimer’s. Epigenomic changes are alterations in the chemical modifications or packaging of DNA that affect the usage of a particular gene within a given cell.
The researchers utilized ATAC-Seq, a technology that analyzes the accessibility of locations across the genome at single-cell resolution, to measure such changes. The researchers were able to combine information about how much a gene is expressed with data on how accessible that gene by merging this data with single-cell RNA-sequencing data. They might also start grouping genes into regulatory circuits that regulate specific cell processes like synaptic transmission, which is the major way neurons relay signals throughout the brain.
Using this method, the researchers were able to follow changes in gene expression and epigenomic accessibility in genes previously linked to Alzheimer’s disease. They also discovered which cells are most likely to express these disease-linked genes, and discovered that many of them are found in microglia, the immune cells responsible for removing debris from the brain.
This study also discovered that as Alzheimer’s disease proceeds, every type of cell in the brain experiences epigenomic erosion, which means that the cells’ typical pattern of accessible genomic locations is lost, contributing to cell identity loss.
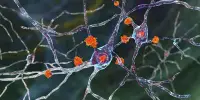
The role of microglia
In a third Cell publication, led by MIT graduate student Na Sun and research scientist Matheus Victor, the researchers focused on microglia, which account for 5 to 10% of brain cells. These immune cells not only clean waste from the brain, but they also respond to injury or infection and help neurons communicate with one another.
Tsai and Kellis’ 2015 publication demonstrated that many of the genome-wide association study (GWAS) variations related with Alzheimer’s disease are mostly active in immune cells like microglia, rather than neurons or other types of brain cells.
The researchers employed RNA sequencing to divide microglia into 12 stages based on hundreds of genes that are expressed at varying amounts during each state in the new study. They also discovered that as Alzheimer’s disease advances, the number of microglia in the brain increases. The Tsai lab has previously demonstrated that as brain inflammation increases, the blood-brain barrier degrades and neurons have trouble interacting with one another.
At the same time, there are fewer microglia in the Alzheimer’s brain, which promotes homeostasis and helps the brain function correctly. The researchers identified transcription factors that activate the genes that keep microglia in that homeostatic state, and the Tsai lab is now investigating ways to activate those factors in the hope of treating Alzheimer’s disease by programming inflammation-inducing microglia to return to a homeostatic state.