Holoprosencephaly
Definition: Holoprosencephaly (HPE) is the failure of the prosencephalon, or forebrain, to develop normally. The forebrain is formed and the face begins to develop in the fifth and sixth weeks of human pregnancy. The condition also occurs in other species. The forebrain is a region of the brain in the fetus that develops into parts of the adult brain, including the cerebral cortex. Instead of the normal complete separation of the left and right halves of the forebrain, there is an abnormal continuity between the two sides.
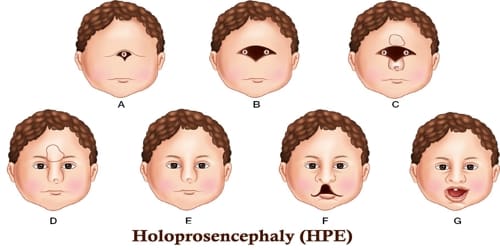
Holoprosencephaly falls into three classes:
- Alobar Holoproencephaly is when the brain does not divide at all. This is usually associated with severe facial deformities.
- Semilobar Holoprosencephaly is when the brain’s hemispheres have somewhat divided, causes an intermediate form of the disorder.
- Lobar Holoprosencephaly is when there is more separation between brain hemispheres. This is the least severe form, and the fetal brain may be nearly normal.
The condition can be mild or severe. Most cases are not compatible with life and result in fetal death in utero.
In less severe cases, babies are born with normal or near-normal brain development and facial deformities that may affect the eyes, nose, and upper lip.
Causes, Sign, and Symptom of Holoprosencephaly: Holoprosencephaly (HPE) is the most common developmental abnormality of the forebrain in humans. It occurs in 1 of every 250 human embryos. Many of these embryos do not survive and are lost to miscarriage. By birth, the prevalence is 1 in 8,000 to 1 in 10,000 live births and stillbirths.
Holoprosencephaly may present with of a spectrum of other malformations and abnormalities. Moderate to severe defects may cause mental retardation, spastic quadriparesis, athetoid movements, endocrine disorders, epilepsy, and other serious conditions; mild brain defects may only cause learning or behavioral problems and possibly mild motor impairments.
Sometimes HPE can be caused by environmental causes. The most common known cause of HPE in humans is maternal type 2 diabetes mellitus. In pregnancies of diabetic mothers, the chance for HPE to occur is about 1%.
Holoprosencephaly (HPE) is a malformation sequence with a very variable degree of severity for both the brain and facial abnormalities. Intellectual disability is associated with HPE and seizures are often present.
Symptoms of holoprosencephaly range from mild (no facial/organ defects, anosmia, or only a single central incisor) to moderate to severe (cyclopia). HPE consists of a spectrum of defects or malformations of the brain and face. At the most severe end of this spectrum are cases involving serious malformations of the brain, malformations so severe that they often cause miscarriage or stillbirth. At the other end of the spectrum are individuals with facial defects which may affect the eyes, nose, and upper lip – and normal or near-normal brain development. Seizures and mental retardation may occur.
Holoprosencephaly (HPE) may also affect other systems in the body. Defects in the pituitary gland can cause an abnormally low level of sugar in the blood (hypoglycemia), low levels of sodium in the blood, or genital abnormalities.
Other genetic syndromes that have been reported in association with holoprosencephaly include Hartfield syndrome (ectrodactyly, cleft lip/palate), agnathia-otocephaly complex (very small chin, ear anomalies), and Pallister-Hall syndrome (extra fingers/toes, hypothalamic hamartoblastomia, anal anomalies).
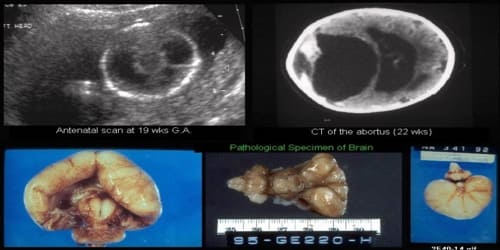
Diagnosis and Treatment of Holoprosencephaly: The diagnosis of holoprosencephaly is usually made by MRI or CT of the brain. Holoprosencephaly can sometimes be detected prenatally through ultrasound or MRI, though mild forms may not be reliably detected prenatally.
Treatment and care for the issues associated with holoprosencephaly are supportive and based on the specific medical issues present for an individual child.
In any case, all individuals who have been found to have some degree of HPE do best if cared for by a team of providers who are coordinated and focus on that individual’s unique needs and set of circumstances. This includes providers that address medical, developmental, and functional needs that a person with HPE typically has and potentially other clinically relevant findings.
In predicting and maximizing the prognosis for a child with HPE, sustained and attentive follow-up by a primary medical provider, whether a geneticist, neonatologist, pediatric neurologist, developmental pediatrician, or primary care pediatrician, is essential to closely follow the child in order to coordinate optimal care and to provide a balanced and realistic prognosis that allows for appropriate treatment, anticipatory guidance, and optimal outcomes.
Information Source: