About Cystic Fibrosis
Definition
Cystic fibrosis or CF is a common grave genetic disease that affects the exocrine glands and is characterized by the production of abnormal secretions, leading to mucus buildup that impairs the pancreas and, secondarily, the intestine. It was affects mostly the lungs, but also the pancreas, liver, kidneys, and intestine. Mucus buildup in lungs can impair respiration. Abbreviated CF. Without treatment, CF results in death for 95 percent of affected children before age 5; however, a few long-lived CF patients have survived past age 60. Early diagnosis is of great importance.

CF is inherited in an autosomal recessive manner. It is caused by the presence of mutations in both copies of the gene for the cystic fibrosis transmembrane conductance regulator (CFTR) protein. It is most common among people of Northern European ancestry and affects about one out of every 3,000 newborns. About one in 25 people is a carrier. It is least common in Africans and Asians.
Treatment includes physical therapy to loosen the mucus in the lungs and use of pancreatic enzymes and medications to fight dangerous infections of the lungs. One in 400 couples is at risk for having children with CF. CF is a recessive trait, so the chance of an at-risk couple having a child with CF is 25 percent with each pregnancy.
Causes, Sign andSymptoms of Cystic Fibrosis
Cystic fibrosis (CF) is an inherited condition. For someone to have CF, they need to inherit the defective gene from both of their parents. The defective gene contains codes for producing a protein that controls the flow of salt and water outside of the organs, including the lungs and the pancreas.
In CF, the balance of salt is disturbed, leading to too little salt and water outside of the cells and the production of thicker-than-normal mucus. It’s caused by a mutation (change) in a single gene called the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR.) This controls the flow of salt and fluids in and out of our cells. If the CFTR gene doesn’t work like it should, sticky mucus builds up throughout our body.
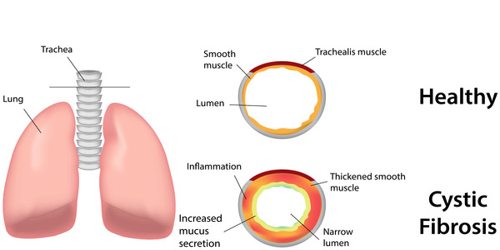
Although most people have two working copies (alleles) of the CFTR gene, only one is needed to prevent cystic fibrosis. CF develops when neither allele can produce a functional CFTR protein. Thus, CF is considered an autosomal recessive disease.
The main signs and symptoms of cystic fibrosis are salty-tasting skin, poor growth, and poor weight gain despite normal food intake, accumulation of thick, sticky mucus, frequent chest infections, and coughing or shortness of breath. Males can be infertile due to congenital absence of the vas deferens.
The most common symptoms of Cystic fibrosis (CF) are:
- salty-tasting skin
- persistent coughing
- shortness of breath
- wheezing
- poor weight gain in spite of excessive appetite
- greasy, bulky stools
- nasal polyps, or small, fleshy growths found in the nose
In rare cases, cystic fibrosis can manifest itself as a coagulation disorder. Vitamin K is normally absorbed from breast milk, formula, and later, solid foods. This absorption is impaired in some cystic fibrosis patients.
Diagnosis and Treatment of Cystic Fibrosis
Cystic fibrosis (CF) is usually diagnosed through a sweat test. Sweat is collected and the amount of chloride, a component of salt in the sweat, is measured. A high level of chloride is an indication of CF.
Genetic tests can also be carried out by analyzing cheek cells or a blood sample. These tests are mainly used to find out if a person carries the CF gene, but they can also be used to confirm a CF diagnosis following an unclear sweat test result. There are over 1,700 known mutations of the CF gene. As a result, most genetic tests for the condition only screen for the most common mutations.
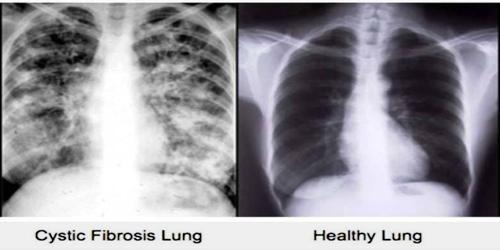
There is currently no cure for CF. Treatment can manage the symptoms of the disease, however, and improve quality of life. Symptoms can vary and treatment plans will be individualized. The most consistent aspect of therapy in CF is limiting and treating the lung damage caused by thick mucus and infection, with the goal of maintaining quality of life. Intravenous, inhaled, and oral antibiotics are used to treat chronic and acute infections. Mechanical devices and inhalation medications are used to alter and clear the thickened mucus. These therapies, while effective, can be extremely time-consuming.
Reference: