Polycystic Liver Disease (PLD)
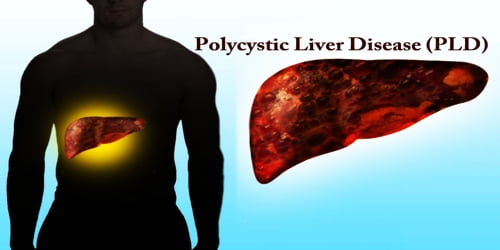
Definition: Polycystic liver disease (PLD or PCLD) is an inherited condition characterized by many cysts of various sizes scattered throughout the liver. It is commonly seen in association with autosomal-dominant polycystic kidney disease, with a prevalence of 1 in 400 to 1000, and accounts for 8–10% of all cases of end-stage renal disease. The much rarer autosomal-dominant polycystic liver disease will progress without any kidney involvement.
People affected by this condition tend to have more and larger cysts as they age and usually start to have symptoms around age 50, although symptoms can begin to occur earlier. However, many affected individuals do not have symptoms. Enlargement of the liver (hepatomegaly) can cause abdominal pain and discomfort, shortness of breath (dyspnea), early satiety and gastro-esophageal reflux. Rare complications are hepatic cyst hemorrhage, infection or rupture.
Polycystic liver disease comes in two forms as autosomal dominant polycystic kidney disease (with kidney cysts) and autosomal dominant polycystic liver disease (liver cysts only). Surgical and medical treatment is available to manage the symptoms, but the only definitive treatment for this condition is liver transplant. Most cases are inherited in an autosomal dominant pattern, but some cases seem to occur with no apparent cause (sporadically).
Sometimes, cysts are found in the liver in association with the presence of autosomal dominant polycystic kidney disease (AD-PKD). In fact, about half of the people who have AD-PKD experience liver cysts. However, kidney cysts are uncommon in those affected by polycystic liver disease.
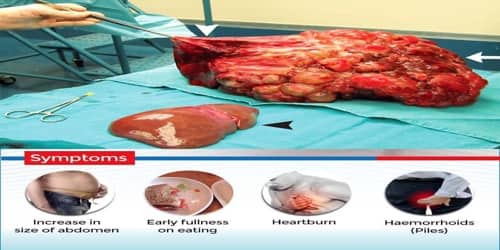
Causes, Signs, and Symptoms of Polycystic liver disease: The majority of people with polycystic liver disease inherit the condition, but PLD can occur randomly with no genetic link. Women are affected by more severe disease compared to men. PLD is most common in people who have polycystic kidney disease (PCKD), with its frequency increasing with age and advanced renal disease.
Most cases of the polycystic liver disease are inherited in an autosomal dominant pattern. Dominant genetic disorders occur when only a single copy of an abnormal gene is necessary to cause a particular disease. The abnormal gene can be inherited from either parent or can be the result of a mutated (changed) gene in the affected individual. The risk of passing the abnormal gene from an affected parent to an offspring is 50% for each pregnancy. The risk is the same for males and females.
Most of the time, people with polycystic liver disease have no symptoms. However, if the liver becomes very enlarged and bulky with cysts, symptoms may include:
- Bloating or swelling in the abdomen
- Abdominal pain
- Feeling full
- Shortness of breath
Only about one out of every 10 people with PLD has problems associated with it. In addition to severe abdominal pain, other complications may include:
- Bleeding into a cyst
- Infection of a cyst
- Bile duct obstruction and jaundice (yellowing of the skin and eyes)
In rare cases, patients can suffer from cyst bleeding (hepatic cyst haemorrhage), or a cyst can be infected by bacteria (hepatic cyst infection), causing pain and fever. Infrequently, large liver cysts may rupture, causing severe abdominal pain. Even with the presence of many cysts, the liver of individuals with polycystic liver disease functions normally.
Diagnosis and Treatment of Polycystic liver disease: Most patients with polycystic liver disease (PLD) are asymptomatic with simple cysts found following routine investigations. After confirming the presence of cysts in the liver, laboratory tests may be ordered to check for liver function including bilirubin, alkaline phosphatase, alanine aminotransferase, and prothrombin time.
It is also possible to test for blood levels of two markers of liver and bile duct disease: gamma-glutamyltransferase (GGT) and alkaline phosphatase (ALP). These two markers might be elevated in patients with severe polycystic liver disease.
Treatment is usually not needed unless people have symptoms. Mild pain associated with PLD can be treated with pain medication. Medication to slow down cyst growth and fluid secretion in the liver (somatostatin analogs, namely octreotide and lanreotide) is also useful in reducing liver volume.
Because they are very expensive, these medications are typically reserved for patients with moderate to severe disease with reduced quality of life.
Many patients are asymptomatic and thus are not candidates for surgery. For patients with pain or complications from the cysts, the goal of treatment is to reduce the size of cysts while protecting the functioning liver parenchyma. Cysts may be removed surgically or by using aspiration sclerotherapy.
In the most severe cases, a liver transplant may be an option. This treatment is typically reserved for people who are experiencing severe abdominal pain, having trouble eating, and whose overall quality of life is suffering. Liver transplant for polycystic liver disease is rarely performed.
Information Source: