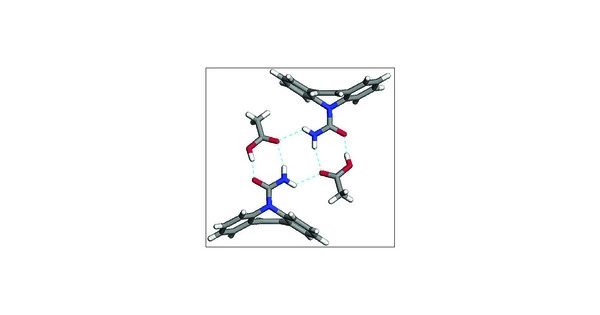
Crystal structure prediction (CSP) is critical in the pharmaceutical industry because a drug’s physical properties can be heavily influenced by its crystal structure. Predicting the most stable and bioavailable crystal structures is frequently used in the discovery and design of new pharmaceutical drugs.
Scientists have rewritten the book on modeling and predicting crystal-free energy. Their research demonstrates that crystal form stability under real-world temperature and humidity conditions can be predicted reliably and affordably using computer simulation.
Physical properties (stability, solubility, etc.) are known to be strongly dependent on solid-state form and environmental factors such as temperature and relative humidity. Recognizing that late-appearing, more stable forms can lead to disappearing polymorphs and potentially market withdrawal of life-saving medicine, the pharmaceutical industry has heavily invested in solid-form screening platforms.
It is not easy to quantify the free energy differences between crystalline forms. Metastable crystal forms can be difficult to prepare in pure form, and they are frequently susceptible to converting to more stable forms. Thus, having the ability to computationally model free energies means that the risks posed by physical instability can be understood and mitigated for all systems, including those that are experimentally intractable.
We owe a fair part of our success to the visionaries among our customers who have enabled us to create an industrial working environment with an academic touch that promotes creativity based on core values such as honesty, integrity, perseverance, team spirit, and genuine care for people and the environment.
Dr. Marcus Neuman
A major impediment to developing computational methods for accurately predicting solid-solid free energy differences has been a lack of reliable experimental benchmark data. There are few reports in the literature, and much of the experimental data on free energy determinations for molecules of pharmaceutical interest is simply not available.
To meet this challenge, academic and industrial experts collaborated to create the first ever reliable experimental benchmark of solid-solid free energy differences for chemically diverse, industrially relevant systems. They then predicted these free energy differences using several methods pioneered by Prof. Alexandre Tkatchenko’s group at the University of Luxembourg’s Department of Physics and Materials Science, and further improved by Dr. Marcus Neumann and his team of researchers at Avant-garde Materials Simulation.
Without using any empirical input, these calculations leveraging high-performance computing (HPC) were able to predict and explain data from seven pharmaceutical companies with surprising accuracy. The potential future implications of this work are manifold, and this latest development is just one of many potential applications of quantum mechanical calculations in the pharmaceutical industry.
“I am thrilled to see how computational methods developed in my academic group have been quickly adopted to reliably predict the energetics of drug crystal forms in the pharmaceutical industry in a matter of years, breaking the traditional barrier between research and industrial innovation,” remarks Prof. Tkatchenko.
“We owe a fair part of our success to the visionaries among our customers who have enabled us to create an industrial working environment with an academic touch that promotes creativity based on core values such as honesty, integrity, perseverance, team spirit, and genuine care for people and the environment,” explains Dr. Marcus Neuman, founder and chief executive officer of AMS, in a statement.
“It is no small feat to build links between fundamental science, high-performance computing, and major industry players in order to make a lasting impact on the future of health,” said Prof. Jens Kreisel, Rector of the University of Luxembourg. “We take very seriously our mission of nurturing an ecosystem where researchers can drive societal change for good.”